US Department of Energy Focuses on Nuclear Physics Inverse Problems: SAGIPS Achieves 80x Efficiency Boost}
The SAGIPS system, developed for solving complex nuclear physics inverse problems, significantly enhances efficiency, demonstrating an 80-fold improvement on large-scale supercomputers.


In recent years, advances in deep generative models have enabled scientists to design therapeutic peptides targeting difficult sites with greater precision. However, they underestimated the critical role of molecular surface interactions in protein-protein interactions (PPI)—like finding the lock but ignoring the right angle to open it—hindering peptide design and discovery.
To address this gap, researchers from the Jefferson National Accelerator Facility led a study in collaboration with Stanford University, introducing the SurfFlow system, a novel surface-based generative algorithm capable of comprehensive peptide design, including sequence, structure, and surface features.
SurfFlow employs a multimodal Conditional Flow Matching (CFM) architecture to learn the distribution of surface geometries and biochemical properties, improving peptide binding accuracy.
In the comprehensive PepMerge benchmark, SurfFlow outperformed all atomic baseline methods across all metrics, demonstrating the advantages of considering molecular surfaces in de novo peptide discovery and the potential of integrating multiple protein modalities for therapeutic peptide discovery.
This research was accepted at the 2025 ACM SIGKDD Conference on Knowledge Discovery and Data Mining (KDD), the most influential international conference in data mining.

Research Background
Peptides, short chains of 2-50 amino acids, play key roles in biological processes like cell signaling, enzyme catalysis, and immune responses. They are essential in pharmacology due to their high affinity and specificity for cell surface receptors, with low toxicity, immunogenicity, and ease of delivery.
Traditional peptide discovery relies on frequent physical energy calculations, but due to the vast design space, this approach is inefficient, prompting rapid development of computational methods.
Recently, the focus on protein-protein interactions (PPI) has highlighted the importance of molecular surface complementarity, including electrostatics, hydrophobicity, and geometric features like protrusions and grooves, which are crucial for specific binding mechanisms like lock-and-key or induced fit.

These surfaces are fundamental interfaces for protein recognition and binding. Therefore, considering sequence, structure, and surface simultaneously during peptide generation enhances the overall design consistency.
SurfFlow
To achieve this, Stanford and Molecular Heart teams developed SurfFlow, a new comprehensive design algorithm applying multimodal flow matching (FM) to internal structures and molecular surfaces, represented by surface point positions and unit normal vectors, within the SE(3) rigid framework.
Since surface complementarity alone does not guarantee successful binding—precise placement of electrostatics, polarity, and hydrophobicity is also necessary—SurfFlow incorporates these biochemical constraints.
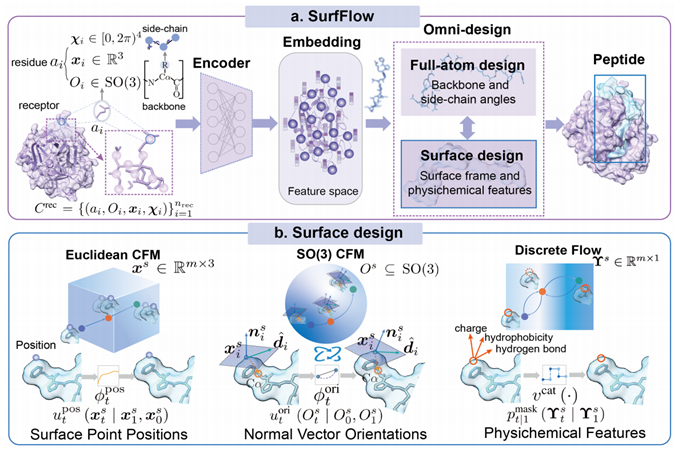
Illustration: SurfFlow workflow for comprehensive peptide design, considering sequence, structure, and surface modality consistency during generation.
It uses Discrete Flow Matching (DFM) for classifying surface features and Continuous-Time Markov Chains (CTMC). To capture irregular surface geometries and multi-scale interactions, an Equivariant Surface Geometry Network (ESGN) models surface heterogeneity and interactions dynamically.
Key features like cyclicity and disulfide bonds, which influence stability and affinity, are included as additional conditions to enhance SurfFlow’s capacity and generalization.
Performance Evaluation
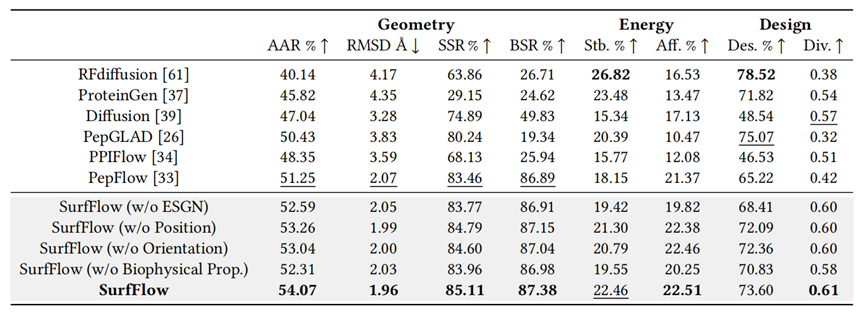
Extensive assessments on the PepMerge dataset from PepBDB and Q-BioLip showed SurfFlow consistently outperformed atomic baselines across all metrics, with ablation studies confirming the importance of its core components.
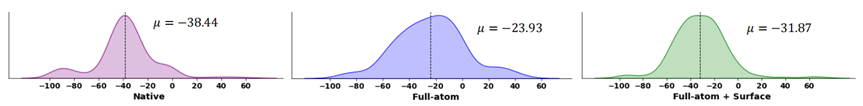
Designs and binding energy distributions for natural and designed peptides, with lower values indicating better binding affinity.
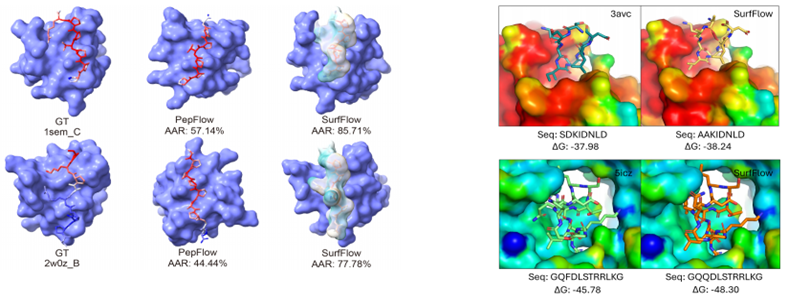
Comparison of peptides designed via DL algorithms and cyclic constraints, demonstrating the impact of structural features on stability and binding.
Although SurfFlow improves upon previous atomic design mechanisms, further exploration is possible, such as incorporating receptor surface information into the joint distribution model. Pretraining on PDB data, as shown by RFDiffusion, could also enhance performance.
Despite these prospects, SurfFlow represents a new model capable of generating all protein modalities—sequence, structure, and surface—simultaneously. The team applied SurfFlow to solve a specific peptide design challenge, integrating cyclicity and disulfide bonds into the generation process.
It is expected that the team will soon release SurfFlow, and interested researchers can look forward to its upcoming availability.