Published in 'Cell', CAS High Cai Xia and Team Develop AiCE: A Universal Strategy for AI Protein Engineering}
CAS researchers introduce AiCE, an AI-informed protein engineering framework integrating structural and evolutionary constraints, enabling rapid, efficient protein evolution without specialized training.


Long-term protein engineering has been limited by low success rates and high costs. An ideal strategy requires minimal effort for optimal performance.
Current AI-based protein engineering methods are computationally intensive, necessitating more user-friendly alternatives that maintain prediction accuracy and promote community adoption.
CAS team led by Gao Cai Xia developed AiCE (AI-informed constraints for protein engineering), which integrates structural and evolutionary constraints into a universal inverse folding model, enabling fast, efficient protein evolution without additional training.
This research, titled Advancing protein evolution with inverse folding models integrating structural and evolutionary constraints, was published in Cell on July 7, 2025.

Link to paper: https://www.cell.com/cell/abstract/S0092-8674(25)00680-4
Inverse folding models
Traditional protein models face challenges like low success rates, high iteration costs, and limited generalization. Deep learning-based methods have achieved success but require extensive computational resources and have limited generalization.
The team observed that generalized inverse folding models like ESM-IF1 and ProteinMPNN, trained on natural protein structures and sequences, can capture complex distributions shaped by evolutionary dynamics. These models can be directly applied without extra training, and recent results show that sampling from inverse folding outputs can identify high-fitness (HF) mutations for antibody evolution, though applicability to larger proteins and complex mutations remains uncertain.
AiCE aims to predict HF single amino acid substitutions by extensive sampling of inverse folding models combined with structural constraints, significantly improving prediction accuracy.
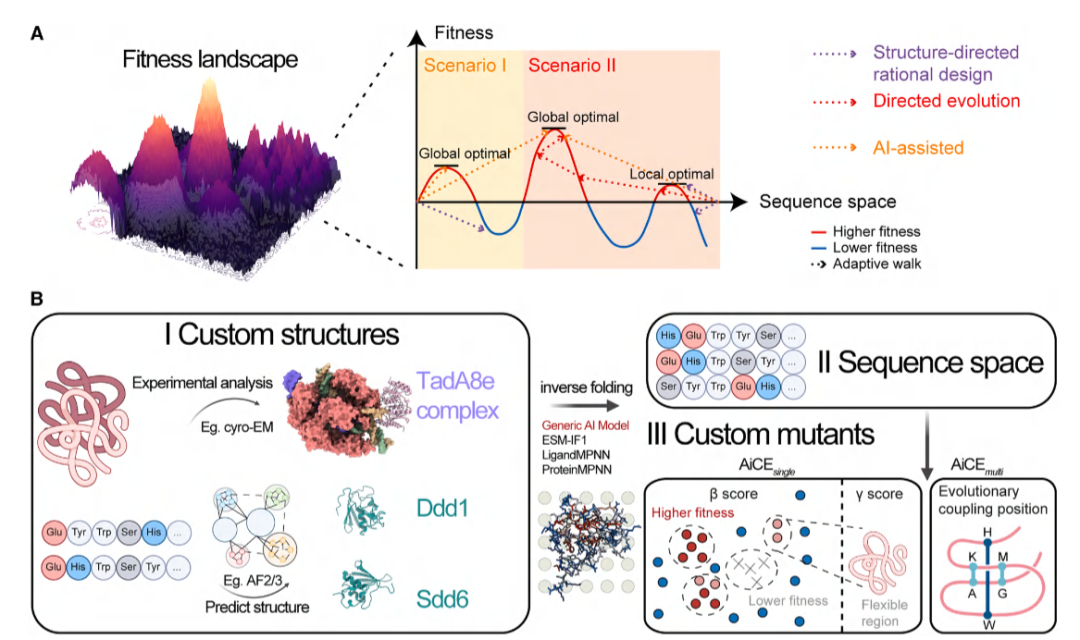
Illustration: AiCE as an AI-driven protein engineering method. (Source: Paper)
AiCE is model-agnostic and can optimize simple protein structures and complex enzymes. Evaluations across eight different protein engineering tasks showed success rates from 11% to 88% for HF mutations.
Based on these results, the team developed precise, efficient base editors, including enABE8e with a smaller editing window, enSdd6-CBE with significantly improved fidelity, and enDdd1-DdCBE with 14.3 times higher mitochondrial editing efficiency.
Summary of achievements
Compared to other methods, AiCE outperformed in 60 deep mutational scanning (DMS) datasets, with performance improvements ranging from 36% to 90%. Its effectiveness in complex proteins and protein-nucleic acid complexes was also validated, with structural constraints boosting accuracy by 37%.
In 31 DMS datasets, the module's ability to identify HF mutations solely through sampling from inverse folding models was evaluated. The results showed high proportions of positive fitness mutations, with accuracy for HF mutations at 12%, and top variants achieving about 47% improvement. AiCE-ProteinMPNN achieved the highest prediction accuracy (35%), outperforming other AI models.
Furthermore, AiCE successfully evolved eight proteins with diverse structures and functions, including deaminases, nuclear localization sequences, nucleases, and reverse transcriptases. These engineered proteins enable the creation of next-generation base editors for precision medicine and molecular breeding.
Era of precise design
AiCE shifts protein engineering from “experience-driven” to “data-and-constraint-driven”—using inverse folding models to explore sequence-structure relationships, enabling efficient design from single to multiple mutations.
AiCE offers a simple, efficient, and broadly applicable protein engineering strategy. By unlocking the potential of existing AI models, it provides a promising new direction for the field and enhances the interpretability of AI-driven protein redesign.
The developed base editors show clinical translational potential, and modifications of nucleases and reverse transcriptases demonstrate cross-scenario applicability. Future work involving molecular dynamics simulations or cryo-EM structural analysis may deepen mechanistic understanding and further improve AiCE.