Microsoft AI4Science Team Develops Orbformer: Say Goodbye to Inaccurate and Unaffordable Multi-Reference Systems}
Microsoft's Orbformer revolutionizes quantum chemistry by enabling accurate, cost-effective wavefunction modeling for diverse molecules, surpassing traditional methods in precision and efficiency.


In molecular and material property simulations, high-precision electronic structure calculations often come with prohibitive computational costs.
Current ab initio methods can accurately describe many-body electronic wavefunctions, but their enormous computational demands limit practical applications.
The complexity is further compounded by the ambiguous boundary between weakly and strongly correlated systems.
For weakly correlated systems (like equilibrium molecules), single-reference methods such as Coupled Cluster (CC) can efficiently and accurately solve wavefunctions. However, these systems are isolated islands in chemical space, with unclear boundaries.
Conversely, for strongly correlated systems (like transition metal compounds or reaction transition states), existing methods lack a unified standard, often requiring manual adjustments by experts, with higher computational costs.
The challenge in computational chemistry is fundamentally a trade-off between accuracy and efficiency.
In this context, Microsoft Research's AI for Science team, in collaboration with researchers from Freie Universität Berlin, has introduced Orbformer—a new paradigm to address these issues.
The related paper, "An ab initio foundation model of wavefunctions that accurately describes chemical bond breaking", has been published on arXiv.

Orbformer is a transferable wavefunction model that can be fine-tuned for unknown molecules, achieving accuracy comparable to classical multi-reference methods at a fraction of the cost. It is the only method that consistently converges to chemical accuracy (1 kcal/mol) on benchmarks and challenging bond dissociation and Diels-Alder reactions.
This work transforms the idea of "sharing the Schrödinger equation solving cost across multiple molecules" into a practical quantum chemistry approach.
Orbformer is suitable for molecules of various sizes, compositions, and geometries, often matching or exceeding the energy accuracy of state-of-the-art single-point methods. It learns composable molecular features and generalizes to new molecules.

Illustration: Pretraining and fine-tuning of Orbformer (source: paper).
Specifically, Orbformer can approximate the electronic Schrödinger equation for various molecules. It takes molecular configuration M and electronic spin coordinates x as inputs.
Using M as the wavefunction network input, rather than as parameters for an optimization problem, is key to sharing calculations among similar molecules.
Orbformer is built from Jastrow factors and generalized Slater determinants, ensuring the wavefunction is antisymmetric with respect to electron exchange for each M.
Its architecture and modules include:

1. Nuclei Message Passing Neural Network (MPNN):
- Input: molecular configuration M
- Outputs: (1) Atomic nucleus feature matrix Q_nf^nuc (for Orbital Generator message passing)
- (2) Rotation matrix W_n^rot (for Electron Transformer’s electron-nucleus features)
2. Dynamic Orbital Generator:
- Includes an additional orbital axis compared to Nuclei MPNN

Input:
- (1) Electron coordinates r_e + nuclear coordinates R_n
- (2) Electron features Tef from Electron Transformer
Output:
- (1) Localized orbital envelopes Ω
- (2) Projected electron representations Φ
Innovations include:
- Learning localized modes via variational principles without preset orbital types
- Exponential decay ensuring negligible long-range interactions, satisfying composability
- Dynamic, transferable parameters for orbitals based on molecular configuration M
3. Electron Transformer for multi-electron correlation modeling (physically interpretable architecture):
- Inputs: electron coordinates and spins x, rotation matrix W_n^rot
- Outputs: electron feature matrix Tef for constructing the generalized Slater matrix
Breakthroughs include:
- Physically aware feature extraction with filters ensuring Kato cusp conditions and size consistency
- Attention mechanisms with distance-dependent bias functions for electron-electron interactions
- Receiver gates to enhance orbital locality and expression capacity

These modules work together to ensure:
- Composability: wavefunction components degrade gracefully during dissociation
- Transferability: orbital parameters dynamically adapt across molecules
- High accuracy in describing bond breaking and transition states via multi-electron correlation
The design emphasizes locality—interactions decay with distance, and orbitals are generated to be local, facilitating reuse in similar chemical environments.
Researchers trained Orbformer to minimize the Hamiltonian expectation value, sampling molecular configurations and electron coordinates from training distributions, extending variational Monte Carlo (VMC).
They created a pretraining dataset of 22,350 molecules with up to 24 electrons, composed of H, Li, B, C, N, O, F in various geometries, optimized through a three-stage training protocol with innovative deep QMC techniques.

Illustration: Orbformer chemical pretraining stages using the Light Atom Curriculum (LAC) dataset (source: paper).
To achieve high-precision wavefunctions, models are fine-tuned from pretraining until convergence, often grouping similar structures and sharing parameters to reduce costs, proportional to the number of geometries.
Performance and Results
Orbformer’s results for transition states in Diels-Alder reactions closely match experimental data.
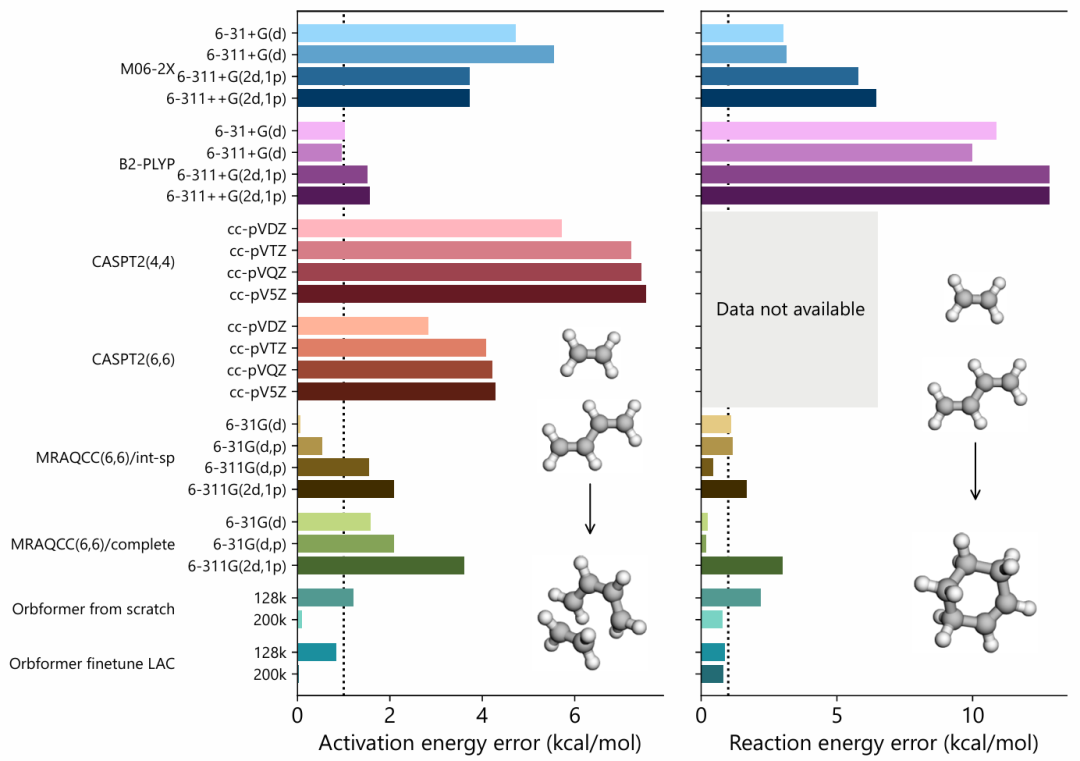
It outperforms classical methods like DFT, NEVPT2, MRCI, and MRCC in cost and accuracy for five-bond dissociation curves, with significant reductions in computational expense and improved precision over early quantum Monte Carlo approaches.
Orbformer directly learns features of the electronic wavefunction, such as localized core orbitals for each carbon atom, without hardcoding these features into the architecture.
Conclusion
Orbformer demonstrates the feasibility of large-scale pretraining and transfer learning for high-accuracy quantum chemistry, offering a more competitive and cost-effective alternative to traditional wavefunction methods.
It leverages learned patterns across molecules and geometries, greatly reducing computational costs and enabling applications like reaction force fields for multi-reference systems.
While not the final story, Orbformer marks a significant step toward building universal models of molecular electronic structure, promising to become a powerful tool in quantum chemistry.
Paper link: https://arxiv.org/abs/2506.19960
Related content: https://mp.weixin.qq.com/s/Dc0RItibTAJ0ooGLQWpEJg