Fudan & Shanghai Jiao Tong Develop Dual-Branch Model, Published in Nature Sub-Journal with 3 Million Cells Processed in 8 Hours}
A novel dual-branch deep learning model by Fudan and Shanghai Jiao Tong efficiently processes 3 million cells in 8 hours, advancing spatial genomics, published in Nature Communications.


In the microscopic world of tissue slices, the spatial distribution of gene expression holds the key to unlocking biological mysteries—how embryos develop livers, why cancer cells invade and metastasize, and more, all depend on where genes are expressed dynamically.
However, traditional spatial genomics methods can only capture gene expression in single slices and cannot identify critical signals of gene spatial position changes under different conditions. (For example, Sepal algorithm’s F1 score for differential spatial expression genes is only 41%)
To address these issues, a team from Fudan University and Shanghai Jiao Tong University proposed the River framework, which uses a dual-branch prediction architecture and post-hoc attribution strategies to rank the contribution of genes (or other features) to condition differences.
This research, titled Prioritizing perturbation-responsive gene patterns using interpretable deep learning, was published in Nature Communications on July 2, 2025.

Why is it difficult to capture gene 'positional changes'?
Understanding the improvements of the River framework over existing models requires first recognizing the experimental challenges faced by traditional omics techniques.
With technological advances, spatial transcriptomics has led to explosive data growth, creating an urgent need for large-scale computational methods to analyze complex gene spatial expression patterns.
Existing methods, such as spatial variable genes (SVG) and non-spatial approaches, fail to identify Differential Spatial Expression Pattern (DSEP) genes. The team developed River to overcome these limitations.
River is an interpretable deep learning method based on the hypothesis that only genes with significant DSEP across slices can help predict slice or condition labels.
In simple terms, River’s process can be summarized as:
1. Designing predictive models to fully utilize spatial gene expression features across multiple slices and conditions;
2. Quantifying each gene’s contribution to the prediction model;
3. Integrating various deep learning attribution methods to prioritize gene patterns.
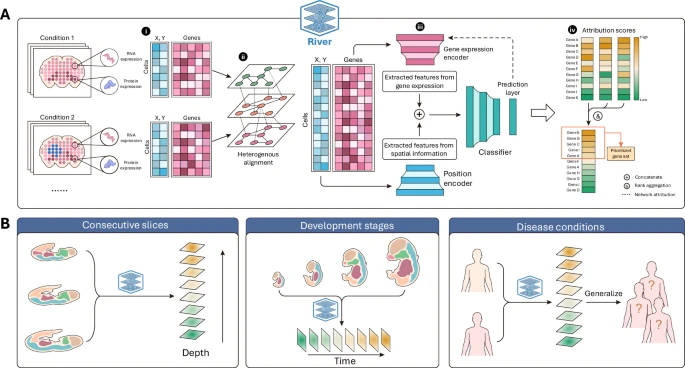
Figure 1: Workflow of River.
The dual-branch prediction architecture includes a positional encoder (to extract features from spatial information) and a gene expression encoder (to extract features from gene data), which independently extract features and fuse them into a latent space.
After training, River uses multiple deep learning attribution strategies to obtain gene contribution scores at the cellular level, then aggregates these scores for a final global ranking.
It emphasizes that the main difference between SVG, Differential Expression Genes (DEG), and DSEP methods is that DSEP focuses on genes with significant spatial distribution changes across conditions, which is crucial in spatial genomics.
Making gene 'positional changes' visible
Of course, the performance of a model must be compared with existing models. In simulated datasets, known perturbed genes are labeled as positive (DSEP genes), while others are negative (background or non-DSEP genes), facilitating evaluation of various methods.
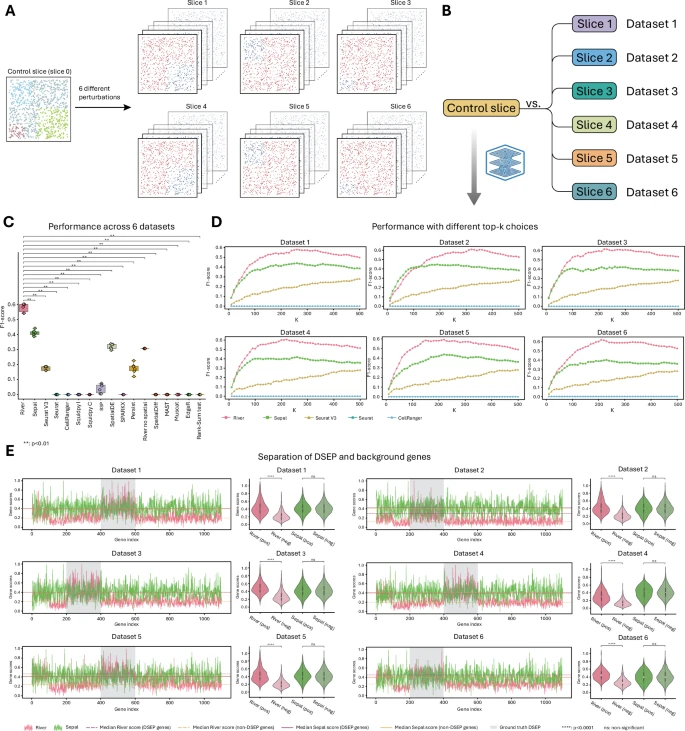
Figure 2: Benchmark comparison.
Performance comparison of River and 16 competing methods (modified to recognize DSEP genes across slices) across six datasets shows River significantly outperforms all others in F1 score (p-value < 0.05), with a median F1 of about 0.59, ranking first.
The second and third are Sepal and SpatialDE, with median scores of approximately 0.41 and 0.32, respectively. The remaining methods are close to zero.
River’s attribution module can assign meaningful scores to each gene, prioritizing those with differential spatial expression patterns. Validation shows River consistently assigns higher scores to true DSEP genes, with significant score differences from background genes.
Tracking gene 'temporal changes'
Most existing studies focus on spatial gene expression within the same slice, neglecting changes over time.
The team applied River to analyze the Stereo-seq dataset of mouse embryos across eight developmental stages. In this context, genes identified as differential may include both spatial and non-spatial changes.
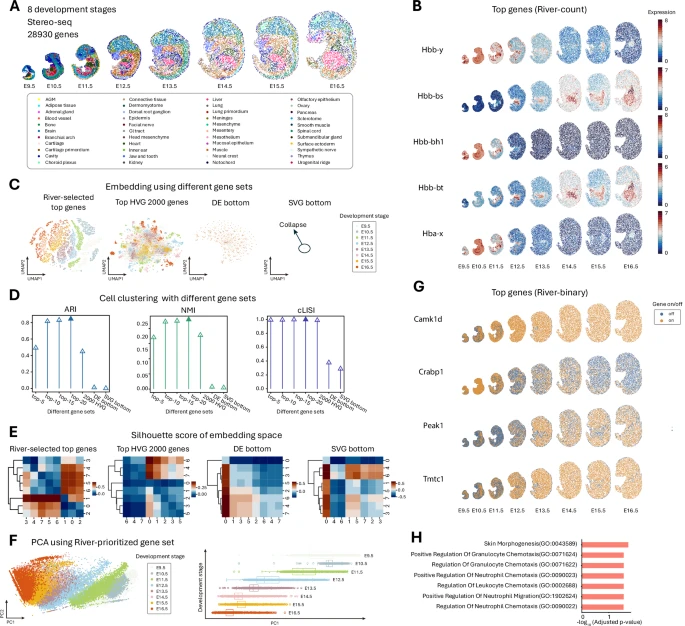
Figure 3: Analysis of mouse embryo across eight developmental stages.
Visualizations confirm that the prioritized genes identified by River show spatiotemporal changes along the developmental axis.
The embedded spatial features effectively distinguish different stages, outperforming the use of 2000 highly variable genes. Conversely, low-priority genes selected by River cannot distinguish stages.
Genes with similar pairwise scores tend to be close in time, showing better clustering, demonstrating River’s ability to capture non-spatial gene expression changes during development.
Practical demonstrations
In real biological scenarios, River also demonstrates strong performance.
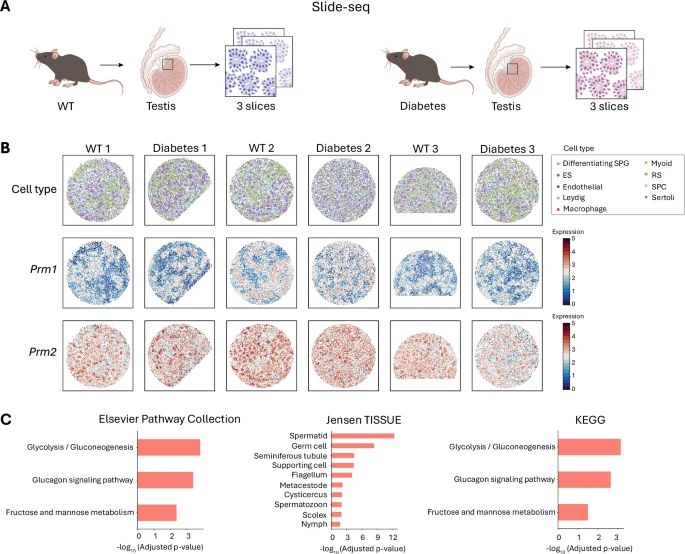
Figure 4: Application on slide-seq dataset.
River can identify DSEP genes involved in spermatogenesis affected by diabetes, as well as genes like Prm1 and Prm2 linked to testicular and embryonic cell loss in diabetes.
It also shows strong generalization ability beyond spatial transcriptomics, demonstrated on the MERSCOPE brain dataset, processing over 70,000 cells in about 7 minutes with three repetitions.
River can process 3 million cells in 5 hours, with near-linear scaling as cell numbers increase. Its large-scale processing capability will become a powerful tool for researchers as datasets grow in size and complexity.
Advancing to a higher level
Results indicate that River is not just a simple differential gene expression or SVG recognition method but is specifically designed to identify DSEPs without limiting assumptions on slices or cell independence.
The emergence of the River framework offers a new perspective for solving differential spatial expression pattern recognition, shifting analysis from static to dynamic spatial pattern parsing.
While results may be influenced by external alignment algorithms, most applications are unaffected, and River can seamlessly integrate with advanced alignment methods to improve performance.
Future research may incorporate contrast modules to strengthen the framework, though batch effects remain a challenge to address.